
One of them is chemical labelling, in which a chemical tagging structure is linked in vitro to the amine terminus (cystein or carbonyl-reactive tags are also available) of the peptides proteomewide via NHS ester reaction. To increase the throughput one can choose, depending on the method, up 11 different isotopically labeled MS tags (TMT distributed by Thermo) or up to 8 different tags for iTRAQ (distributed by ABSciex).
The major problem with these labeling strategies are co-isolated and co-fragmented peptides and their isotopes, which going to get isolated within the precursor selection window and ending up in the specific TMT or iTRAQ MS2 report ion channel. This mixed reporter ion channel is what protein scientists call ratio distortion.
Recently two approaches got introduced to the scientific community to solve this.
1.) EASI, which is a amine reactive TMT series utilizing sulfoxide chemisty during fragmentation. The major advantages is that EASI TMT comes off at lower collisonal energy and before the peptide backbone breaks apart, so you going to have a less complex MS2 spectra at some point.
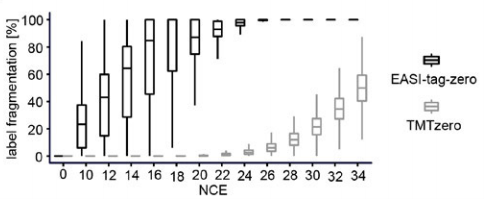
The tricky thing here is to find an optimized setup in which the stepped normalized collisional energies provide enough fragments for determination of the peptide sequence while not losing sensitivity from your EASI reporter ions. More details you will find on the paper by Winter et al 2018.
https://www.biorxiv.org/content/early/2017/11/27/225649.full.pdf+html
The other technological improvement is called MultiNotch MS3 and was introduced by Gygi Lab from Havard in 2014 (its a bit late but I discovered it just now)
When using MultiNotch scan mode for a TMT labeled bottom-up proteomics samples MS duty cycle starts with a high resolution MS1 followed by precursor selection by the quadrupole plus mild CID fragmentation within the low resolution IT , followed by (now the magic comes into play) collection of multiple MS2 precursor with within the multipole (Thermo markets this a SPS which is basically nothing else then multiplexing on the MS2 level) and consecutive HCD to induce reporter ion generation. These fragments are finally scaned with a high resolution orbitrap mass analyis. Thats quite a duty cycle. It would be nice know the time dimensions of this impressive duty cycle!?

It is important to mention that this is only possible with unique instrumentation which allows you to apply multiple fragmentation techniques to the different levels of precusor. So that you are able to get rid of everything which breaks easily in CID and transfer your peptides entirely and still fully tagged to the MS3 stage. Since you discarded all unnecessary ions from your spectra, sensitivity is enhanced by 10-fold compared to MS2 HCD for TMT quantification. The only instrumental platform on which you can utilize such an approach is the orbitrap lumos.
Awesome technology - I love it!
Keine Kommentare:
Kommentar veröffentlichen